


原因は興奮しすぎだった!-320x180.jpg)














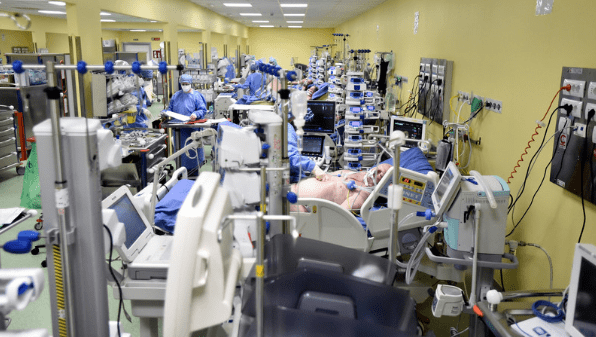




あわら市のジェネリック医薬品メーカー「小林化工」は、水虫薬などに睡眠導入剤を混入したことによる健康被害が相次いだため、医薬品の製造・販売許可を取り下げて4月1日から事業を停止することになりました。
同社は、未承認の手順で医薬品を製造した後、問題のある医薬品の回収やデータの不正が発覚していたが、県からの中止命令後も経営再建に至らなかった。同社は昨年3月、工場や設備をジェネリック大手「沢井グループホールディングス」(大阪市)の子会社に譲渡。4月1日付で医薬品製造販売業の許可を取り下げ、医薬品の公定価格を撤廃し、承認整理を行うことを決定した。
撤退とはいえ、同社製品によって健康被害を受けた人への補償は継続するとのことです。
日本における医薬品製造販売承認の導入について
日本は世界でも有数の医薬品市場であり、1億2600万人以上の人口を抱え、革新的な医薬品に対する高い需要があります。しかし、日本の医薬品に関する規制の枠組みは独特で複雑であり、外国企業にとっては難解なものとなっています。
独立行政法人医薬品医療機器総合機構(PMDA)は、日本における医薬品および医療機器の安全性、有効性、品質を監督する責任を負う規制当局です。PMDAは、医薬品が必要な基準を満たしていることを確認するために、製造販売承認申請の審査と製造販売後調査を行う役割を担っています。
日本で医薬品を製造・販売しようとする企業は、日本の規制要件に従わなければなりません。これを怠ると、法的な問題や風評被害、医薬品ライセンスの取り消しなどが発生する可能性があります。
日本における医薬品の製造・販売に関する規制の枠組みについて

日本における医薬品の製造・販売に関する規制の枠組みは、薬事法(PAA)および薬事法施行規則によって規定されています。これらの規則は、医薬品の製造販売承認を得るための要件や、医薬品の製造販売許可を維持するための条件について定めています。
日本で医薬品の販売承認を得るためには、企業はPMDAに申請書を提出する必要があります。申請書には、製品の安全性、有効性、品質に関するデータ、製造工程や表示に関する情報などを記載する必要があります。
PMDAは申請書を審査し、製造工程が必要な基準を満たしていることを確認するため、現地調査を実施します。製品が安全で有効であると判断された場合、PMDAは販売承認を与えます。
日本における医薬品の製造販売承認取得のための留意点

日本での医薬品の販売承認取得には、時間とコストがかかります。企業は、規制要件を満たし、必要なデータや情報をすべて提供する必要があります。
重要な検討事項のひとつは、日本での臨床試験の必要性です。企業は、製品の販売承認を得るために、日本で臨床試験を実施する必要があります。治験はGCP(Good Clinical Practice)ガイドラインに従って実施され、PMDAの要件を満たさなければならない。
また、企業は製造工程が必要な基準を満たしていることを確認する必要があります。PMDAは、製造工程が規制に適合していることを確認するために、現地調査を実施します。企業は、製造工程と品質管理措置に関する詳細な情報を提供する必要があります。
最後に、企業はラベルが規制に適合していることを確認しなければなりません。ラベルには、製品の安全性、有効性、品質に関する情報、推奨される用法・用量に関する情報が含まれていなければなりません。
日本における医薬品の承認プロセスにおける共通の課題

日本での医薬品承認プロセスは複雑で、外国企業にとっては困難なものです。よくある問題のひとつに、日本での臨床試験の要件があります。日本での臨床試験の実施には時間と費用がかかるため、企業はPMDAの要件を満たすようにしなければなりません。
もう一つの問題は、言葉の壁です。すべての文書は日本語で作成されなければならず、企業は規制プロセスをナビゲートするために言語を十分に理解する必要があります。
最後に、企業は製品が日本独自の規制要件を満たしていることを確認する必要があります。PMDAは安全性、有効性、品質について厳しい基準を設けており、企業は自社製品がこれらの基準を満たすことを確認しなければなりません。
日本における医薬品の製造販売承認を維持する。
医薬品が日本で販売承認を取得した後、企業は医薬品ライセンスを確実に維持しなければなりません。これを怠ると、ライセンスの取り消しや法的問題に発展する可能性があります。
企業は、PMDAが定める市販後要件を遵守しなければなりません。これには、製品が要求される基準を引き続き満たしていることを確認するための市販後調査の実施も含まれます。また、企業は、副作用をPMDAに報告しなければなりません。
さらに、企業は、製造プロセスが要求される基準を継続的に満たしていることを確認しなければなりません。PMDAは、製造プロセスが規制を遵守していることを確認するために、定期的な現場検査を実施することがあります。
日本における医薬品ライセンス保護の取り消し条件の理解

PMDAは、企業が規制要件を満たさない場合、日本での医薬品ライセンスを取り消す権限を持っています。医薬品ライセンスが取り消される条件には、以下のようなものがあります:
- PMDAが定める製造販売後要件を遵守していないこと
- PMDAへの副作用報告の不履行
- 安全性、有効性、品質に関する要求基準を満たさないこと
- 日本国内における医薬品の製造・販売に関する規制違反
日本における医薬品ライセンスの剥奪を回避するための戦略について

日本における医薬品ライセンスの取り消しを回避するために、企業は規制を遵守し、必要な基準を満たすようにしなければなりません。これには、市販後調査の実施、副作用の報告、製造工程が必要な基準を満たしていることの確認などが含まれます。
また、企業はPMDAとオープンなコミュニケーションを保ち、情報提供や現場視察の要請があった場合には、速やかに対応するようにしなければなりません。
最後に、企業は日本における規制要件を十分に理解し、その分野の専門家から助言を得ることが必要です。
日本における医薬品製造・販売承認の複雑な世界を乗り切るためのベストプラクティス

日本における医薬品の製造と販売承認の複雑な世界を乗り切るために、企業は以下のことを行う必要があります:
- その分野の専門家にアドバイスを求める
- 日本における規制要件について十分に理解していることを確認する。
- PMDAが定める市販後の要求事項を遵守すること。
- 製造販売後調査の実施とPMDAへの副作用の報告
- PMDAとオープンなコミュニケーションを保ち、情報提供や現場視察の要請があれば迅速に対応する。
- 製造工程が必要な基準を満たしていることを確認する。
- 日本語をよく理解している
結論から言います:医薬品製造販売業者にとって、日本の規制の徹底的な理解が重要な理由。

アジア市場での事業拡大を目指す医薬品メーカーや流通業者にとって、日本の規制を十分に理解することは不可欠です。日本における医薬品に関する規制の枠組みは独特で複雑であり、規制の遵守を怠ると法的問題や風評被害が発生する可能性があります。
企業は、PMDAが定める市販後要件を確実に遵守し、市販後調査を実施し、副作用をPMDAに報告する必要があります。また、PMDAとのオープンなコミュニケーションを維持し、製造工程が要求される基準を満たすようにしなければなりません。
これらのベストプラクティスを実践し、専門家のアドバイスを受けることで、企業は日本における医薬品製造・販売承認の複雑な世界を乗り切り、製品が安全性、有効性、品質に関する要求基準を満たすことを確実にすることができます。






コメント